
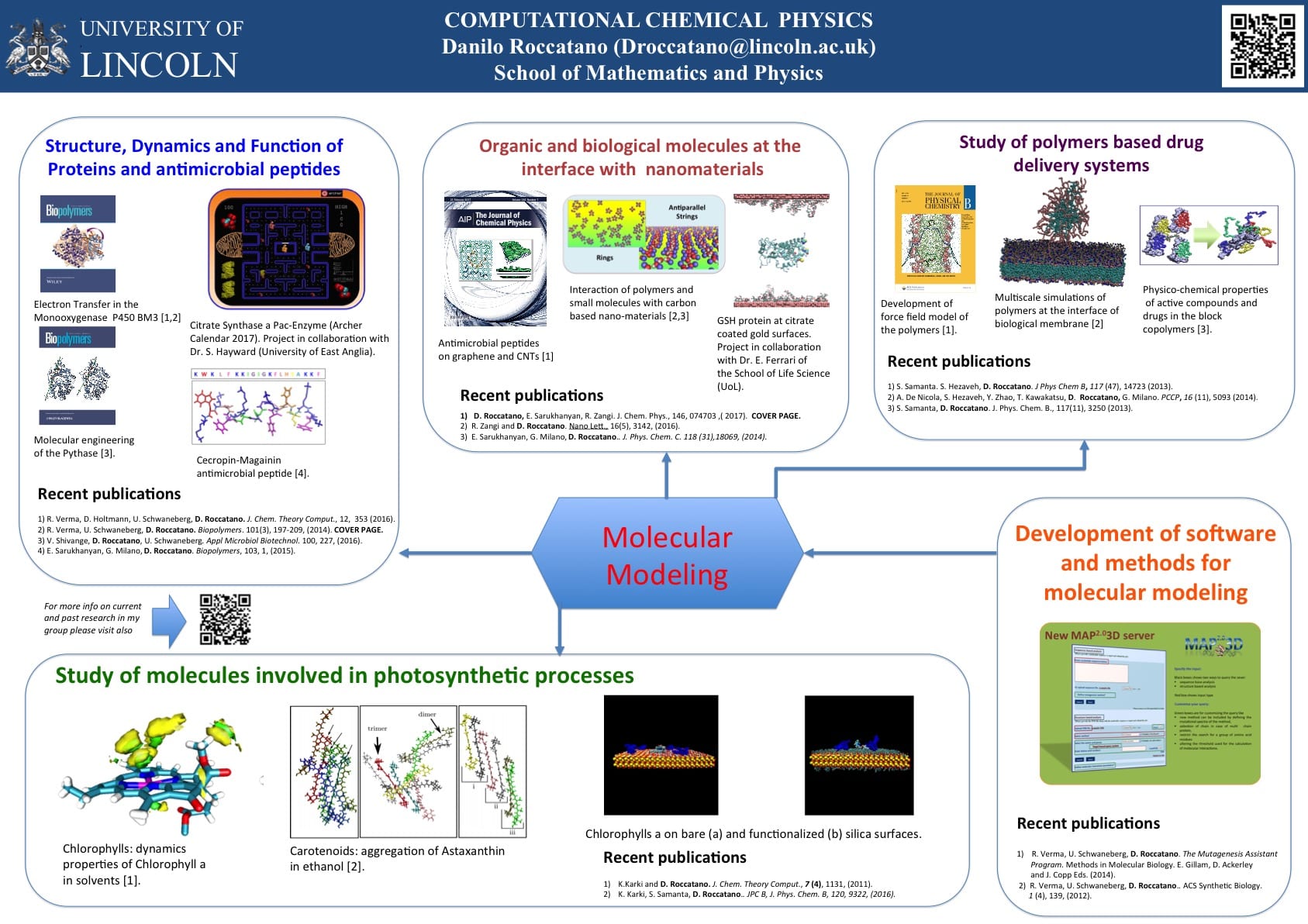
My current research activity comprises different areas of computational chemistry, biophysics, and bioinformatics. I apply and develop computational methods to understand their structure, dynamics, and function of molecular systems. The aim of my research is both the understanding of the modus operandi of these systems at the molecular level and, eventually, their improvements for possible technological or biomedical applications. My future scientific interests are summarized as follows. The citation numbers refer to the list of Publications.
Computational nanomaterials and nanoscience for nanomedicine
The understanding of interactions of biocompatible polymers with nanostructured materials has important technological applications in industry and in medicine. In particular, block copolymers are broadly used for nanotechnological and biomedical applications. Despite this common usage, details on interaction mechanism of polymers with these systems are not yet completely understood at the molecular level. My future research aims to study, using both atomistic and coarse-grained computer simulations, the interactions of nanostructured soft materials with biological interfaces and novel nanomaterials. These studies can disclose molecular interaction mechanisms between biomolecules and scaffolding polymeric materials with possible impacts in molecular medicine and biotechnology. In particular, my researches in this area are directed in two major directions.
-
Multi-scale molecular dynamics study complex hybrid polymers/biological materials
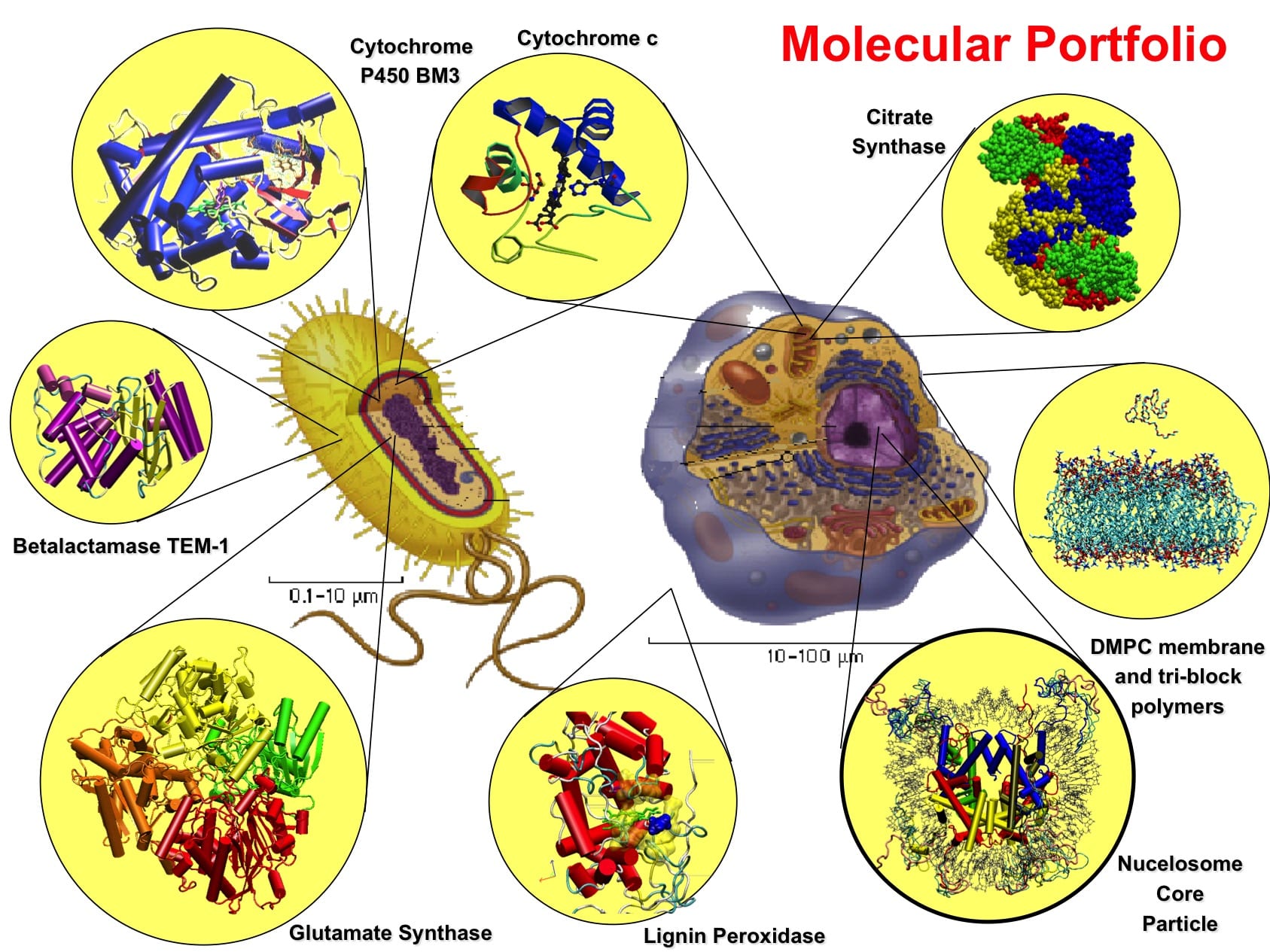
The use of polymeric materials as drug delivery systems is an active area of research with large impact in medicine. Polymeric scaffolds are used to prevent rapid turnover of easily degradable and poorly soluble drugs by substantially increasing their biocompatibility and solubility. The conjugation or encapsulation of therapeutically or diagnostically relevant molecules in polymers micelle or nanoparticles reduce their enzymatic or chemical degradation and other metabolic excretory processes. As a consequence, the residence time, bioavailability, and the transport to target body tissues will improve as well as the effectiveness of the drug. The improved transport capability is important for therapeutics applications requiring the drug diffusion through the tightly packed barriers present in human body. In the past decades, polyether based amphiphiles have increased their significance as drug delivery nanomaterials since their peculiar physicochemical properties can be easily tailored for specific needs. They are currently used as the delivery system of important active molecules against cancer maladies and other life-threatening diseases. Although these materials have been studied for many years, a detailed description of their interactions with the biological material at the atomic level has not yet clarified. For this reason, I am interested studying these systems at the molecular level using molecular dynamics (MD) methods at the multi-scale level to gain a better understanding of these processes. In an initial project, supported by the German research-funding agency (DFG), we have developed accurate models of biocompatible linear poloxamers to study their interactions with molecular interfaces. The results of these studies have revealed interesting details of the interaction mechanism with lipid bilayers and small molecules [15,21,22,24,8]. I am currently extending these studies to other biocompatible polymers of different morphology (e.g. poloxamine and polyglycerols). I am particularly interested studying their interaction properties of these materials with biological membranes, proteins, antimicrobial peptides (AMP), and natural products and drugs. AMP is considered one of the possible new remedies against the increasing spreading of antibiotics-resistant bacteria. Therefore there is a huge interest in AMP since they can be the possible candidate for novel antibiotics. In this area, I have already studied co-solvent mediated structure stabilization mechanisms of different AMP peptides (as Melittin [74, 58] and CA-MA [1] peptide) in solutions. Therefore, I am planning to extend these researchers and analyze the effect of this environment on the structure and function of AMP peptides.
The use of polymeric scaffolds is also used to reduce the degradation of the molecule by the proteases present in the blood. Arginine deiminase (ADI) is a tetrameric arginine-degrading enzyme that catalyzes the irreversible hydrolysis of L-arginine to citrulline and ammonia. ADI based depletion is a target-specific therapy for arginine-auxotrophic tumors such as hepatocellular carcinomas (HCCs), and melanomas. Pegylated ADI is currently under clinical trials for HCC with promising results. In collaboration with the protein engineering group of Prof Schwaneberg at RWTH Aachen, we have studied mutants of the recombinant ADI with improved catalytic activity [31,32]. We are also studying the dynamics of this large enzyme, and the substrate diffusion in the catalytic site. I am planning to study the interaction of different variants of the enzymes conjugated with the polyethylene glycol at the atomistic and coarse-grained level to understand the effect of the polymer on its dynamics and ligand binding in the active site. These studies will help to understand the role of the different catalytic site residues on the substrate specificity of the enzyme and to improve its antitumor drug use.
Others aspect of large interest related to protein is the possibility to use membrane channels to device release systems for polymeric nanocontainers [35]. Specialized transmembrane bacterial proteins can be easily incorporated into polymeric membrane providing a ready-to-use nano valve for bioengineering purpose. So far three bacterial transmembrane channel proteins (OmpF, Tsx, and FhuA) have been used for this scope. The hybrid protein/polymer nanocontainer is called Synthosome. Despite the experimental progress in the engineering of these systems, many aspects of their properties remain at atomic level unsolved and wait for direct investigation using theoretical and computational tools. The theoretical analysis of these hybrid system offers a challenging new perspective to combine techniques from computational biology, physics, and chemistry in order to clarify a large variety of problems related to these apparently simple systems. The nature of polymer/protein interactions, the formation of inclusion body in polymeric curved films, diffusion of solute from nanocontainer with a protein-based gate, are the main aspects that this project will address. The level of scale of these processes varies over several orders of magnitude and for this reason, the study cannot be afforded fully atomistic but a multiscale approach is required. A hybrid atomistic/coarse-grained simulations method permits to study processes having different and independent time and space scale in an efficient manner.
Finally, I am also studying different small organic molecules of nanotechnological and biomedical interest such as Curcumin [14], Chlorophylls [33], Carotenoids, boron clusters [21]. In particular, I am interested studying their aggregative properties in solution and in the presence of polymeric environments. For these compounds, it would be particularly useful to calculate spectroscopic properties since they can be more easily measured experimentally. In this direction, I am working on combining atomistic MD simulations with QM calculations to account the environmental effect on the absorption spectra [55].
-
Computational study of carbon-based nanomaterials
Carbon nanomaterials, as the hollow carbon nanotubes (CNT) and the planar graphene nanosheets (GNS), own many peculiar properties, which makes them attractive for applications in nanotechnology and in the emerging field of nanomedicine. It has been shown that CNTs can act as ion channel blockers, artificial muscles, sensors, as well as, drug-delivery vehicles. In addition, the addition of molecular anchors on these surfaces, with tailored properties, opens the way to create functional interfaces for specific applications. One example is the possibility to create antibacterial surfaces for medical use by coating the surface of the hybrid material with antimicrobial peptides. For these applications, it important to understand at the molecular level the interaction mechanisms of these novel materials with peptides and cellular membranes. In this direction, my researches are focused in two areas of research. I am particular interested in these two aspects.
- Study of interaction mechanisms of small molecules with molecular interfaces. This is a more fundamental aspect of my research. Many dynamics aspects of self-organization of molecules confined on the surface on in the hollows cavities of nanomaterials are still little known. In this direction, I am investigating the self-structuring properties of small solvent molecules, AMP peptides (e.g. Ref. [1]), biologically active molecules (e.g. curcumin [11], porphyrins [30], chlorophylls, carotenoids) and ions [18] in solution and at the interface of different carbon-based nanomaterials to understand their peculiar properties in different conditions.
- Absorption and permeation of functionalized carbon-based nanomaterials into the cell membrane. This part is an extension of the previous one to more complex materials. So far, we have investigated the interaction of single and multi-walled carbon nanotubes with biological membranes and linear polyether polymers and peptides [2,1,3,4,23,25]. The current and future work in this direction is integrated with my first research focus direction on the properties of biocompatible polymers. In this sense, I am particularly interested understanding the following processes: 1) the binding and functionalization of carbon-based nanomaterials by these soft materials; 2) the adsorption mechanism and dynamics of AMP, proteins and small molecule on functionalized graphene and nanotube surfaces; 3) the binding and percolation of these hybrid nanomaterials thought biological membranes.
Molecular biochemistry and biophysics
I have studied using MD simulations structure dynamics and functions of different prokaryotic and eukaryotic proteins.
More recently I have focused the attention on proteins of relevant interest for biotechnological and medical applications. I am interested studying the effect of temperature, co-solvents, and ions on their structural and dynamics properties. I am particular interest to understand these effects on the following systems.
- Recently, it has been shown that it is possible to modify metalloenzymes, such as monooxygenases, to introduce new functionalities (Nature 2016, DOI: 1038/nature17968 ). These new discover pay the way to new class of tuneable green catalyst for cutting-edge applications in synthetic chemistry. One possible scaffold for these new enzymes is the well-studied P450BM-3 monooxygenase from Bacillus Megaterium that catalyses the oxidation of a vast number of hydrophobic compounds from alkanes to steroids and fatty acids. Its ability to insert with high specificity and selectivity an oxygen atom into an inactivated C-H bond makes it a very attractive enzyme for biotechnological applications. The possible replacement of iron with other metals could potentially extent its capability to other class of compounds. In collaboration with U. Schwaneberg of RWTH of Aachen (Germany), I have studied this enzyme for several years by analysing different functional aspects of this enzyme by comparing crystallographic and mutagenesis data with computer simulation and molecular modelling of the Heme domain (the catalytic domain) of the enzyme [56,52,59,42,38,37,18,10]. Recently, we have also used MD simulations to investigate the effect of interdomain conformational changes and dynamics of the FMN/heme domains complex and its effect on the interdomain electron transfer (ET) mechanism [6]. These simulations evidenced an inter-domain conformational rearrangement that reduces the average distance between the FMN and heme cofactors in agreement with previously proposed hypotheses suggesting that the crystallographic FMN/heme complex is not in the optimal arrangement for favourable ET rate under physiological conditions. The estimation of the electron transfer rate, using semi-empirical calculation of the electron tunnelling, in the protein during the simulation shown the occurrence of ET pathways between the two redox centres with calculated ET rates comparable with the experimental one. In addition, the analysis of the collective modes of the protein complex evidenced a clear correlation of the first two essential modes with the activation of the most effective ET pathways along the trajectory. We have also studied the interaction of the same domains with the alternative electron mediator Cobalt (II) Sepulchrate [13] that can substitute the natural and expensive electron source, NADPH, to make more viable biotechnological exploitation of the enzyme. This approach permitted to correlate the identified ET pathways to the protein dynamics.
My future directions are aimed to understand the function of the complete enzyme. For this purpose, I aim to build a reliable model that satisfies all biochemical and biophysical data available. In this sense, modeling studies are directed to combine both coarse-grained and atomistic MD simulations to explore the possible complex structure of this elusive structure. The electron pathway analysis combined with the available mutagenesis data will be used as the guideline for testing the reliability of the models.
My interest on the metalloenzymes go beyond the P450 BM3, and I am also interested studying computational approach to design active site scaffolds of others type of enzymes with substituted metal centers that can perform abiotic catalysis (Science 2016, DOI: 10.1126/science.aah4427). These studies will be integrated with the developing of the MAP3D tool described in the last paragraph.
- I am also studying extremophiles proteins to understand at the molecular level the temperature effect on their properties. I am intrigued about the subtle relation between dynamics and functions that in these molecules play a fundamental but still not clear relation. I have recently focused my attention to the proteases that have several applications in diagnostics and industrial products and thereby requiring a high resistance towards temperature, chaotropic salts and detergents like guanidinium chloride (GdmCl) and sodium dodecyl sulfate (SDS). We have studied the structure and dynamics at different temperature of the psychrophilic protease subtilisin S41 from the Antarctic bacillus TA41, and two variants with two and seven amino acid substitutions [30]. These results of the study supported the hypothesis that the introduced amino acid substitutions, rather than improving the global stability of the variants by increasing its rigidity, lead to a change of the principal fluxional modes allowing the protein to explore a different subset of conformations. A better understanding of this process can open alternative strategies to increase the enzyme stability in addition to increasing the rigidity of the protein scaffold. In another study, Subtilisin E, a well-studied serine protease, was reengineered by directed evolution and rational design into a chaophilic (high resistance to GdmCl and SDS denaturants) protease for applications in diagnostic kits [7, 21].
Currently, I am studying in collaboration with Dr. S. Hayward (University of Norwich, UK) the mechanism of domain motion in different thermal adapted citrate synthase (CS) [63,76]. In particular, we are using umbrella-sampling methods to characterize the thermodynamics of the substrate binding and release of the product. Our aim is the understanding the molecular adaptation of the dynamics and thermodynamics properties of the different thermal adapted CS’s at a quantitative level.
Computer-aided protein engineering and structural bioinformatics
I have developed a web-based tool called MAP (Mutagenesis Assistant Program) [48,49] (online since the beginning of 2006) to help in designing protein engineering experiments. This tool has been used to explore the properties of genetic code in relation to directed evolution experiments [17,29,39,41,46,47]. The most recent release of the server (MAP2.03D: http://map.jacobs-university.de) [8,20] can analyze also the 3D structure of the protein and correlated with the results of the standard MAP analysis of the protein sequence.
A new version of the server is currently under development and it will incorporate features to improve the representation of the results, the analysis of protein structures and the simulation of protein random mutagenesis experiments.